Agosto es el mes de la concienciación sobre la Atrofia Muscular Espinal #AME. Una enfermedad Neuromuscular de origen genético, en la que se van perdiendo progresivamente neuronas motoras del asta anterior de la médula espinal y los núcleos del tronco encefálico, produciendo atrofia y debilidad muscular simétrica progresiva.
Aunque su inicio y gravedad es variable (Figura 1), la mayoría de los pacientes presentan fenotipos graves (AME Tipo I y II) y fallecen de forma precoz o necesitan soporte respiratorio permanente a partir de los 2 años aproximadamente.
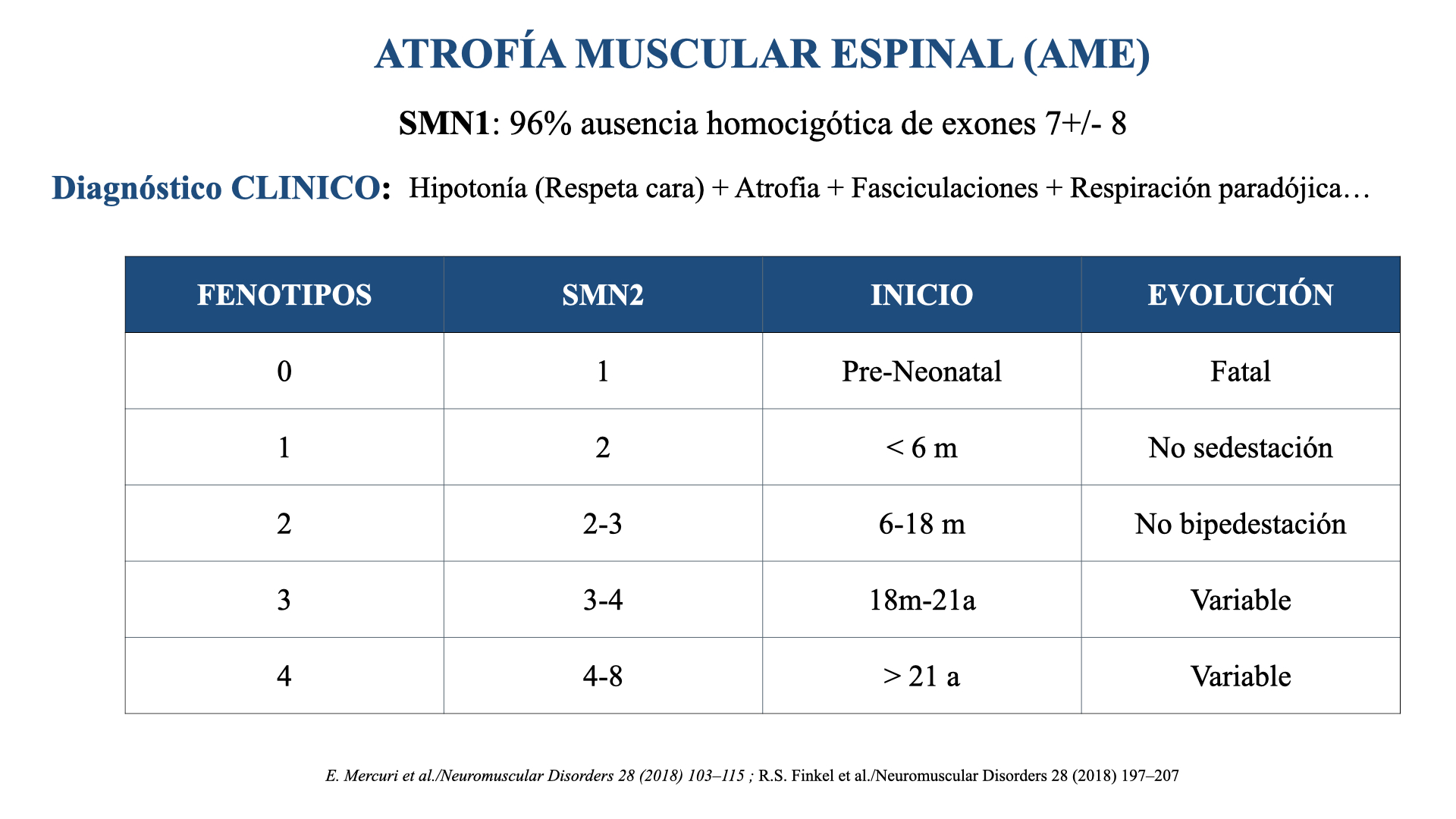
Figura 1. Formas clínicas de AME según el número de copias, el inicio de la clínica y el pronóstico.
Sin embargo, la aparición de nuevos tratamientos y su aprobación para la atrofia muscular espinal (AME), han supuesto un antes y un después en el tratamiento de las enfermedades neuromusulares, convirtiendo una enfermedad mortal como el AME tipo I, en una condición tratable.
Por ello, y aunque se ha avanzado mucho en la comprensión, el diagnóstico y el tratamiento, acortar los tiempos de diagnóstico sigue siendo un reto.
“Tiempo es músculo y calidad de Vida”
Puesto que los pacientes con AME tipo 1 tienen una pérdida irreversible de neuronas motoras antes de nacer, que progresa dramáticamente hasta la perdida prácticamente completa en torno a los 6 meses de vida, el retraso diagnóstico, aunque común, es una catástrofe.
Cuanto más tiempo tardemos en iniciar el tratamiento, peor será el pronóstico de estos niños (Figura 2)
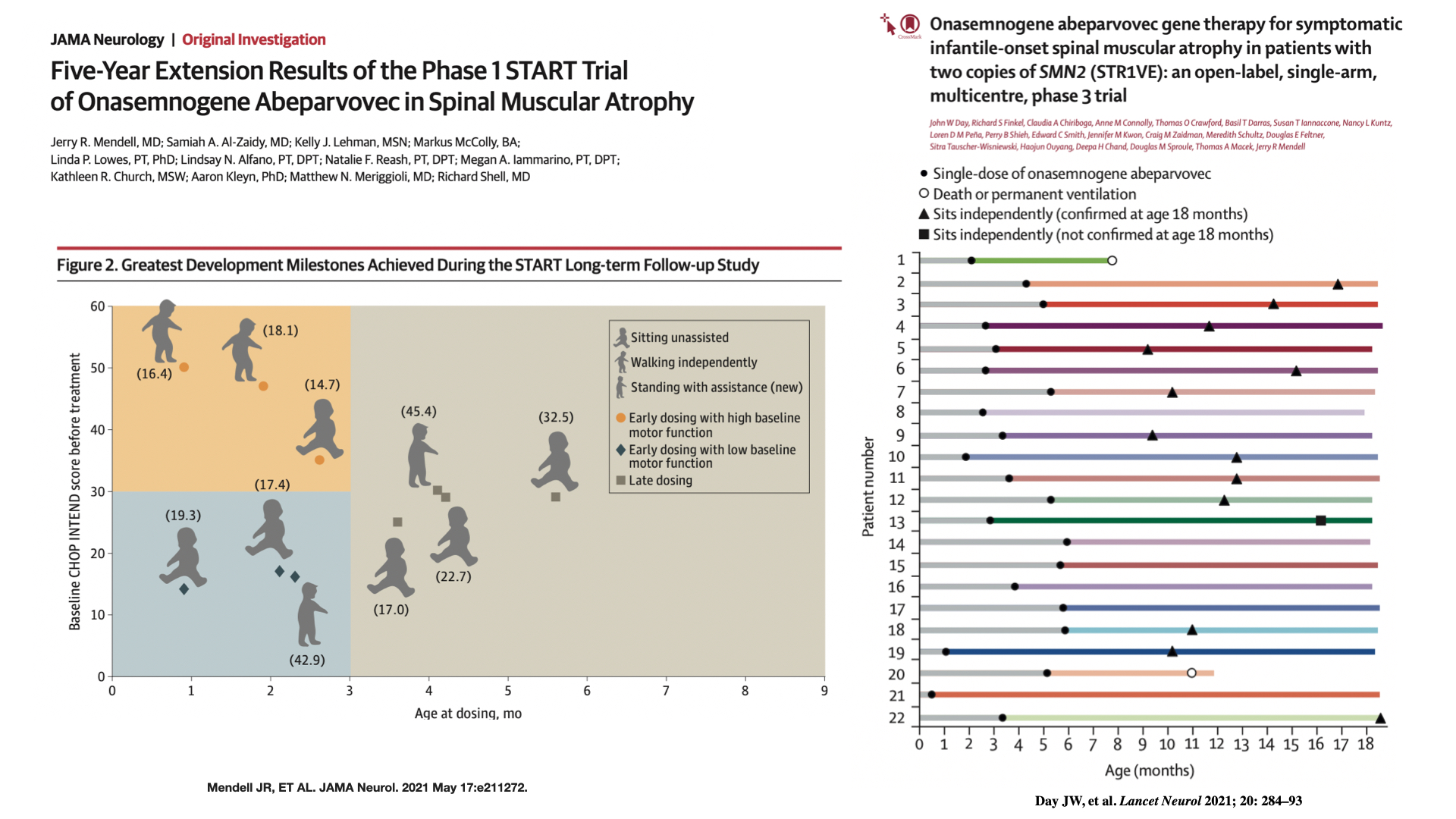
Figura 2. Resultados del tratamiento según la edad de inicio.
Aunque los primeros síntomas se inician en torno a los 2,5, 8 y 39 meses en el Tipos 1, 2 y 3 respectivamente, el diagnóstico de AME no se establece hasta varios meses después (Figura 3).
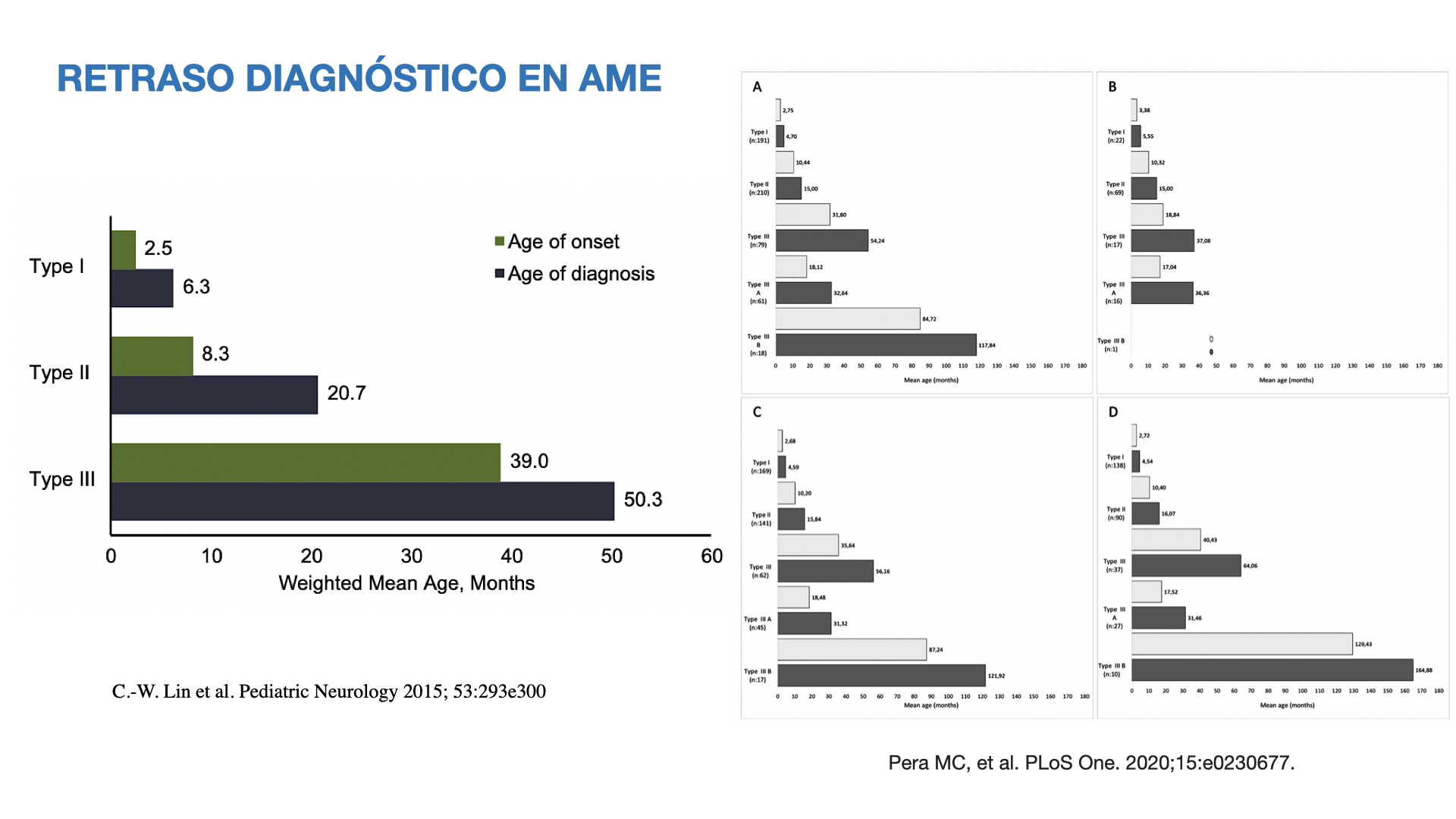
Figura 3. Diferencia de Tiempo entre la aparición de los primeros síntomas y el diagnóstico según el Tipo de AME.
¿Qué podemos hacer para acortar esos tiempos?
1. Conocer los principales Signos de Alarma
http://https://youtu.be/Pb5tCLxi4H4
De forma general podríamos decir que desde el punto de vista motor, son claros signos de alarma:
a. Mal control de la cabeza y movimientos globales pobres, con dificultad para levantar las piernas contra la gravedad entre los 3-6 MESES.
b. No haber adquirido la capacidad para sentarse a los 9 MESES.
c. Incapacidad para ponerse en pie con apoyo a los 12 MESES:
d. No haber adquirido la capacidad para caminar a los 18 MESES.Además de estos signos motores, es frecuente, sobre todo en los casos mas graves la presencia de:
– Fasciculaciones en la lengua
– Respiración anormal (Abdominal)
– Dificultades en la alimentación (problemas para tragar)
– Temblor distal…etcLos primeros signos clínicos que suelen identificar los padres o médicos según el tipo de AME, se detallan en la Figura 4.
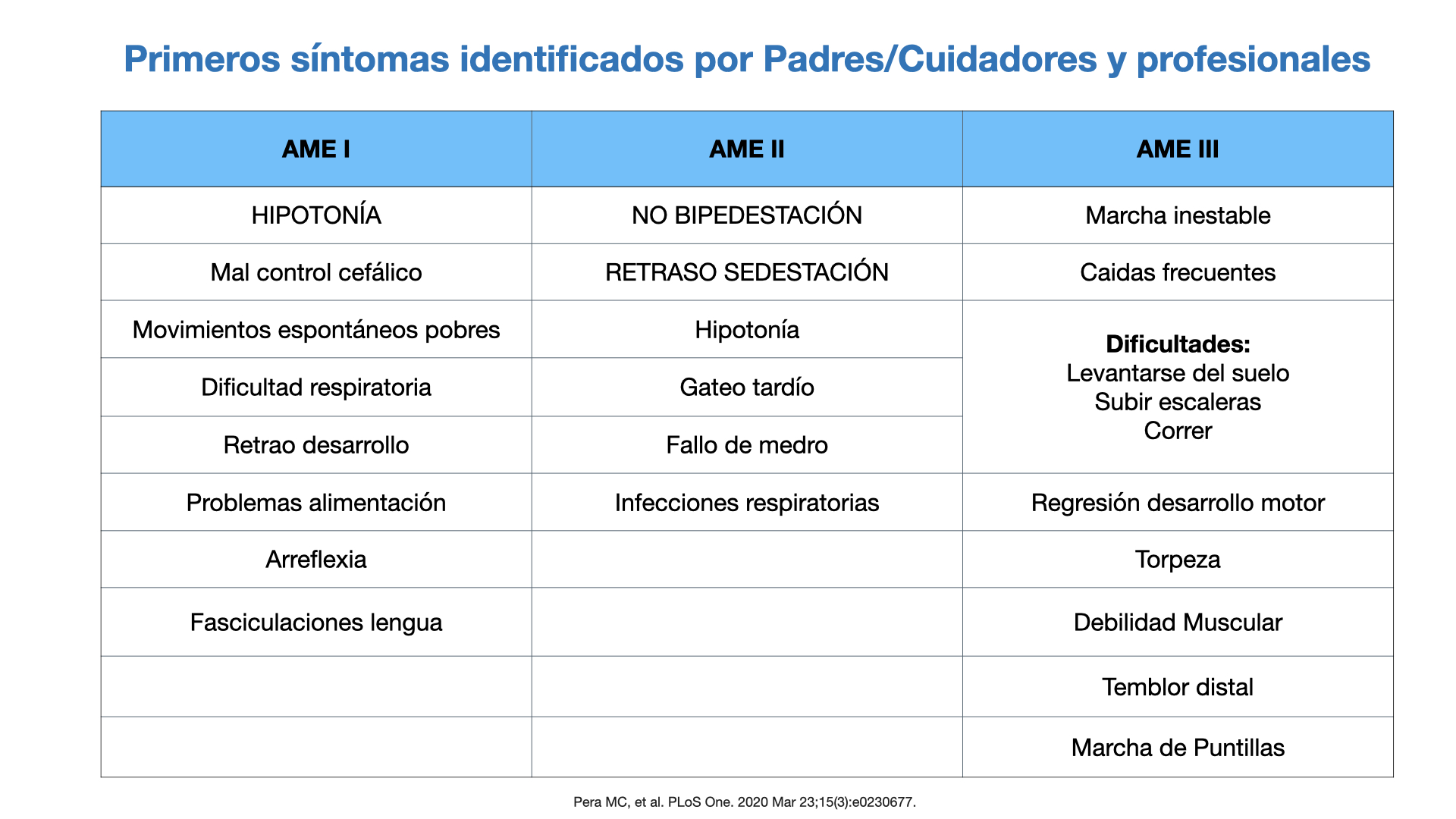
Figura 4. Primeros signos y síntomas de AME
2. Cribado Neonatal
Puesto que el resultado de los nuevos tratamientos es mejor, cuanto más temprano se inicia el tratamiento, el cribado neonatal debería ser considerado como el mejor instrumento para evitar la odisea diagnóstica.
Por ello, aunque podría pasar por alto algunos casos de AME (por ejemplo, aquellos con una mutación puntual) o plantear ciertos conflictos (como tratar pacientes con AME Tipo 3 que no presentarán síntomas hasta varios años después o aquellos con progresión lenta), el cribado neonatal parece la mejor opción.
Además, la AME cumple todos los criterios para ser incluida dentro del screening neonatal (Figura 5).
Figura5. Criterios para Cribado neonatal.
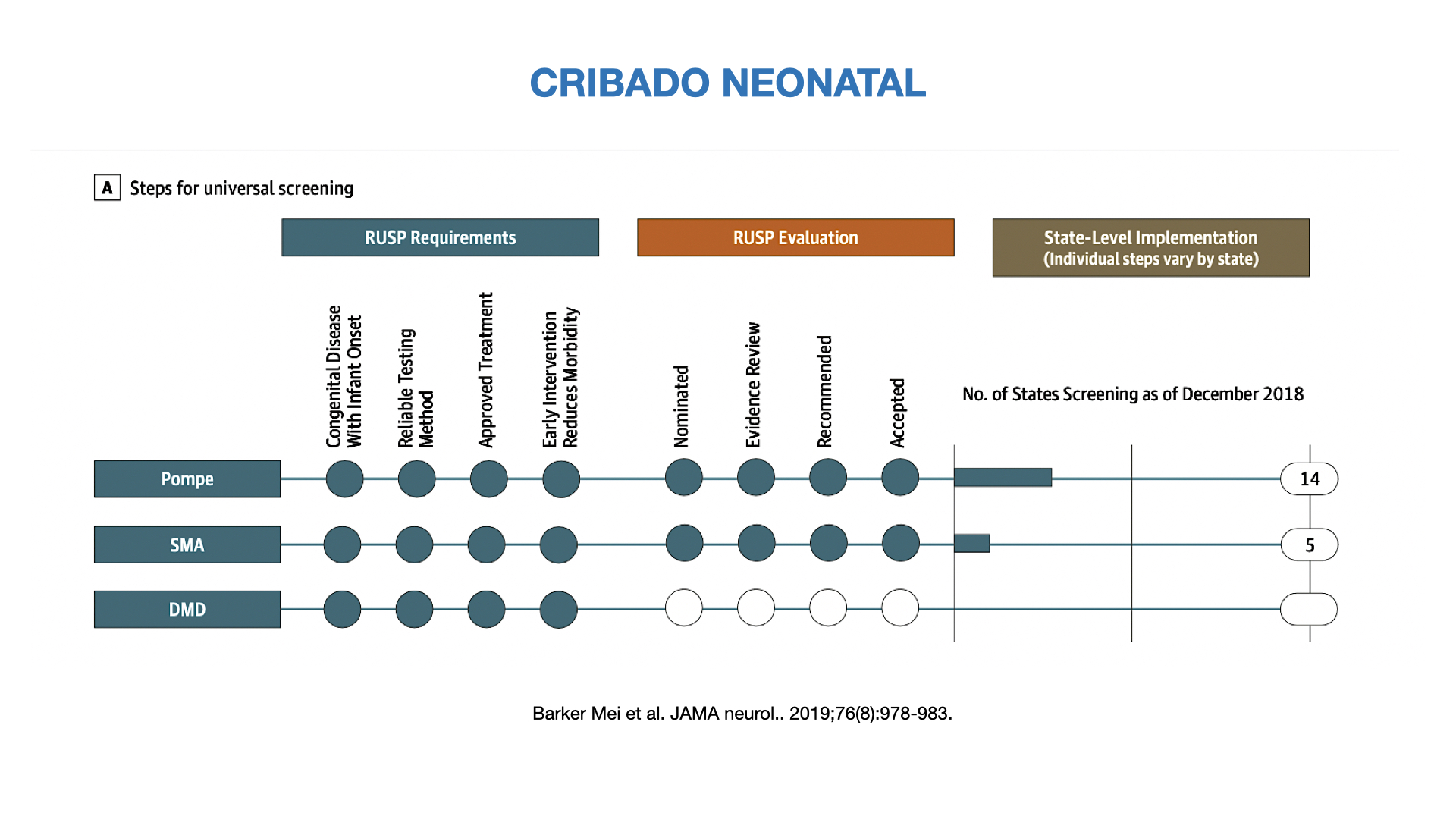
Figura 6. Cribado neonantal en neuromuscular
¿Qué opciones terapéuticas existen?
La piedra angular del tratamiento se basa en un diagnóstico precoz y un abordaje multidisciplinar (Figura 7).
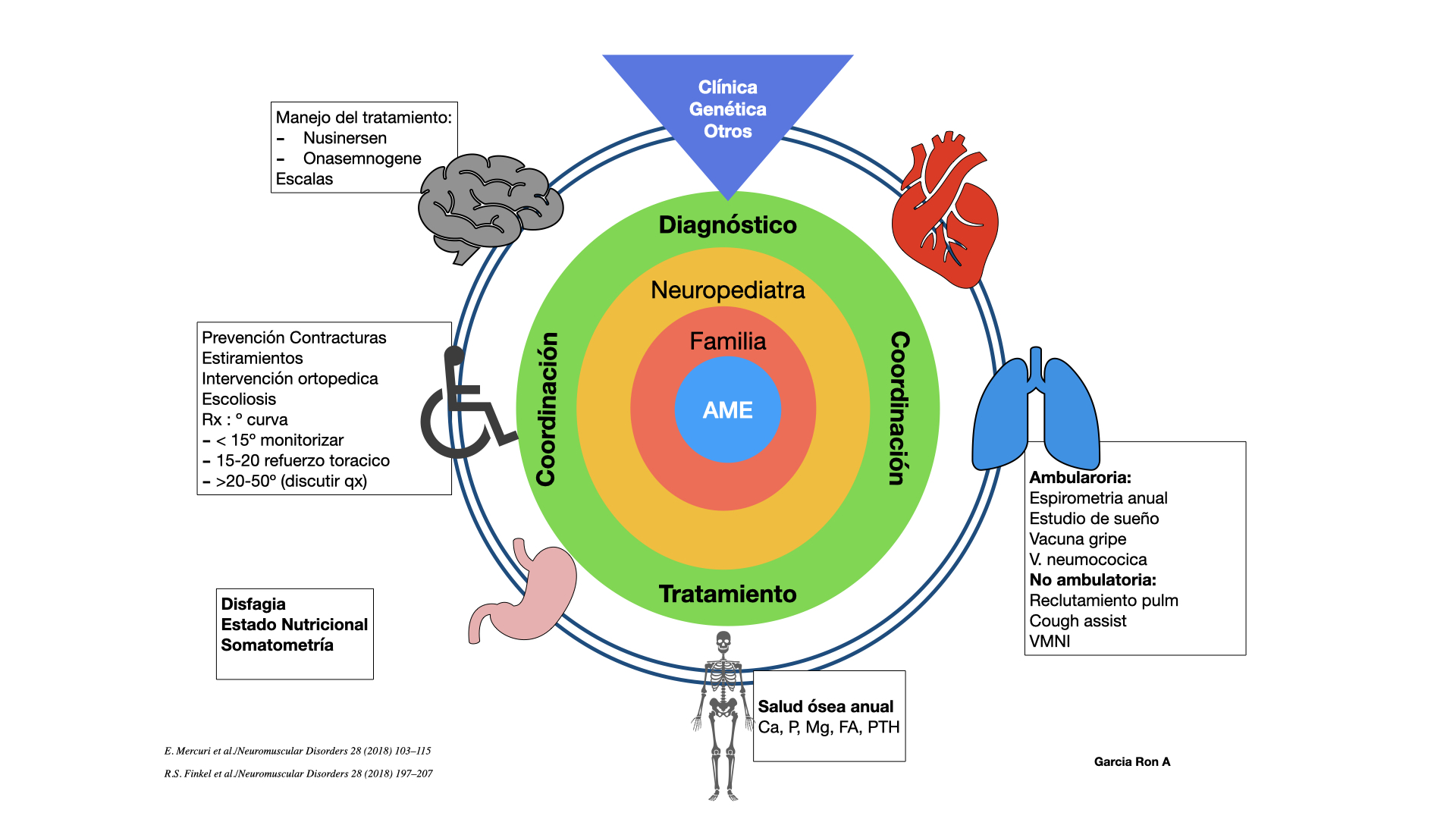
Figura 7. Tratamiento multidisciplinar en AME
Además, actualmente disponemos de tratamientos específicos aprobados por la Agencia Europea del Medicamento (EMA) que van dirigidos a la causa subyacente de la enfermedad (medicina de precisión).
CONCLUSIONES
Aunque hemos avanzado mucho en la comprensión, el diagnóstico y el tratamiento de la AME desde su primera descripción en 1891, su alta incidencia y el beneficio de una intervención temprana hacen imprescindible el diagnóstico precoz. La AME cumple todos los criterios y debería considerarse su inclusión en los programas de cribado neonatal. Mediante una prueba genética sencilla podría confirmarse en pocos días el 95% de los casos, y el escenario terapéutico cambiaría, dirigiéndose a pacientes presintomáticos.
Hasta que esto suceda, debemos formarnos, conocer las signos de alarma del desarrollo psicomotor, y concienciar a los pediatras de atención primaria de la importancia de derivar de forma precoz a estos niños a unidades especializadas para el diagnostico.
Si os ha resultado interesante, no olvideis compartir.
Podéis dejar vuestras preguntas rellenando el formulario
Muchas gracias.
Colaboración con Novartis Gene Therapies
 adrian garcía ron
adrian garcía ron 
Compartir esta entrada